
IGV 是常用的基因組瀏覽工具之一,但在項目交付與論文級出圖中,其在批量制圖、統一風格和自動化方面存在一定局限。
pyGenomeTracks 采用命令行與配置文件驅動,更適合:
l多樣本統一風格對比
l批量輸出指定區域圖片
l高分辨率論文與報告配圖
因此,pyGenomeTracks 常被作為 IGV 的有效補充或替代方案。
在一些生信分析項目中,將測序信號直觀地展示在基因組上,是用戶最容易理解、有說服力的結果形式之一。
本文將系統介紹 pyGenomeTracks 常用的軟件命令及關鍵參數,幫助讀者理解基因組可視化圖像是如何生成的。
一、為什么要做基因組信號可視化?
在各類基于測序的基因組學實驗中(如 ChIP-seq、ATAC-seq、DAP-seq 等),我們的核心目標,都是在全基因組尺度上觀察某個因子或染色質特征“出現在哪里、強不強"。
如果只用表格或一串數值來展示結果,這些信息往往顯得抽象而零散:
峰值有多少、富集倍數是多少,看起來“很科學",但并不直觀,也不利于快速判斷實驗到底做得好不好。
而基因組信號可視化,正是把這些數據“畫到染色體上"。
通過將測序信號轉化為沿染色體連續變化的曲線,我們可以一眼看到:
l目標因子是否在啟動子、增強子等特定區域明顯富集
l不同重復樣本之間的信號形態是否一致、穩定
l實驗樣本與 Input 或背景對照之間,是否存在清晰可辨的差異
這種“看圖說話"的方式,不僅能幫助我們快速評估實驗質量,也為后續的峰值篩選、功能注釋和生物學解讀提供了直觀可靠的依據。
二、本案例使用的軟件工具
本流程主要使用 pyGenomeTracks 軟件包中的兩個工具:
1. make_tracks_file:用于根據已有結果文件,自動生成可視化所需的配置文件。
2. pyGenomeTracks:用于讀取配置文件,在指定的基因組區域內繪制信號圖像。
三、軟件獲取與安裝
推薦使用 Conda 安裝 pyGenomeTracks:
conda install -c bioconda pygenometracks -y
安裝完成后可通過以下命令檢查:
lpyGenomeTracks --help
lmake_tracks_file --help
四、輸入數據文件說明
1.BigWig 文件(信號文件)
BigWig 文件用于存儲測序 reads 在基因組各位置的覆蓋強度或歸一化信號值,是繪制基因組信號曲線的基礎數據形式。
相比原始的 BAM 文件,BigWig 體積更小、讀取速度更快,尤其適合用于基因組瀏覽器或繪圖工具進行連續信號展示。
在本案例中,BigWig 文件包括:
lS1.bw:實驗樣本重復 1 的基因組信號
lS2.bw:實驗樣本重復 2 的基因組信號
lInput.bw:Input 對照樣本
用于評估背景噪音和非特異性信號通過同時展示多個 BigWig 文件,可以直觀比較不同重復之間的一致性,以及實驗樣本與對照之間的差異。
2.BED 文件(區域文件)
BED 文件用于標記基因組上的離散功能區域,例如 peak、結合位點或候選調控區域。
與 BigWig 所展示的連續信號不同,BED 文件更關注“哪里發生了顯著事件"。
在本案例中,使用 peak_location.bed 文件來展示預測得到的轉錄因子結合區域。
將 BED 區域疊加到信號軌跡上,可以幫助我們判斷:
lpeak 是否對應真實、穩定的信號
l富集信號是否集中于少數明確的區域,還是分散且噪音較大
l不同樣本中同一peak區域的信號表現是否一致
3.GTF 文件(注釋文件)
GTF 文件用于提供基因組結構注釋信息,包括基因、轉錄本、外顯子、內含子以及啟動子等功能單元的位置。在基因組信號可視化中,GTF 文件的作用主要體現在兩個方面:
l為信號和 peak 提供生物學背景,幫助判斷其位于基因的哪個結構區域
l解讀信號的潛在功能意義,例如是否偏向啟動子區域或基因間區
通過將 GTF 注釋與 BigWig 信號、BED 區域同時展示,可以實現從“信號存在"到“信號有什么生物學含義"的過渡。
五、自動生成 tracks.ini 配置文件(軟件命令)
在實際分析中,不需要手工編寫復雜的配置文件,而是通過以下命令自動生成:make_tracks_file --trackFiles S1.bw S2.bw Input.bw peak_location.bed sample.gff3 --out tracks.ini
該步驟的作用可以理解為:告訴軟件需要展示哪些文件,以及每個文件在圖中作為一條獨立軌道顯示。
生成的 tracks.ini 文件中已經包含了軌道名稱、顏色、高度等基礎設置。
六、統一信號顯示范圍(常用參數說明)
在默認情況下,每個 BigWig 信號軌道會根據自身數據自動縮放顯示范圍。在多樣本對比時,這種方式容易造成視覺上的誤解。因此,通常會人為設定統一的信號顯示上限。
七、繪制基因組信號圖(核心命令)
完成配置文件修改后,即可使用以下命令繪制基因組信號圖:
pyGenomeTracks --tracks tracks.ini --region NC_083379.1:10813150-10833150 -o image.png --dpi 300
參數解釋:
ltracks:指定軌道配置文件。
lregion:指定繪制的基因組區域,格式為“染色體:起始位置-終止位置"。
lo:輸出圖片文件名。
ldpi:輸出圖片分辨率,300 dpi 適合用于項目報告和論文。
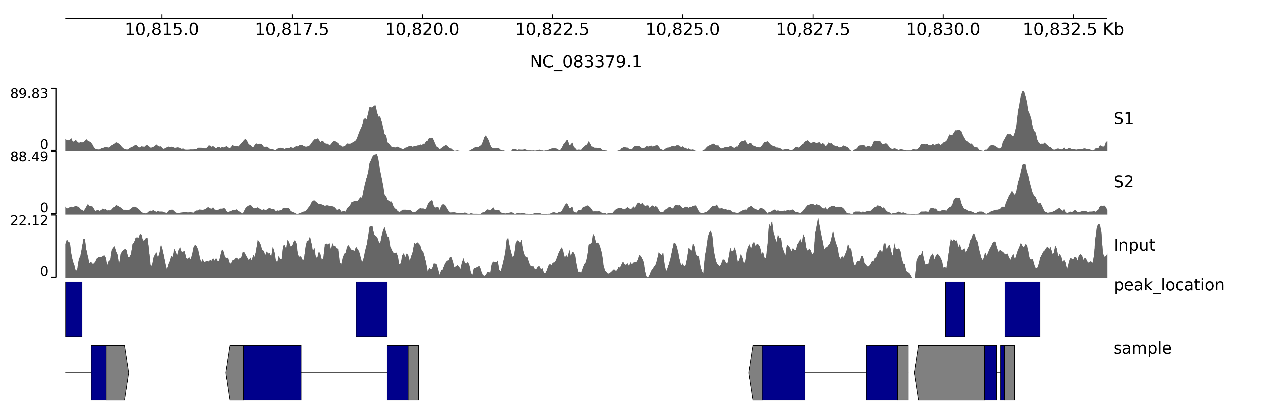
圖 1 默認配置示例圖
八、tracks.ini 中常關注的參數
在配置文件中,以下參數對圖像理解重要:
ltitle:軌道名稱,用于區分不同樣本
lheight:軌道高度,避免圖像過于擁擠
lcolor:軌道顯示顏色這些參數直接影響圖像的清晰度和可讀性。
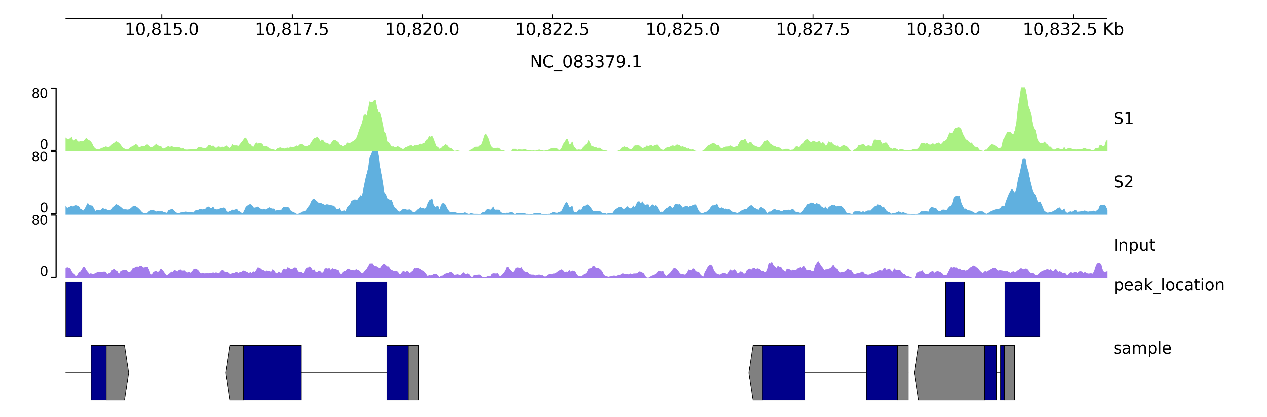
圖 2 修改配置文件示例圖
九、可以從結果圖中獲得哪些信息?
通過最終生成的基因組信號圖,用戶可以直觀看到:
l目標區域是否存在明顯富集
l不同重復樣本之間的信號一致性
lInput 和 IGG 等是否呈現低背景
lpeak 是否與高信號區域對應
這些都是判斷實驗是否成功和結果是否可靠的重要依據。
附錄:pyGenomeTracks 可視化配置參數完整對照表
(一)坐標軸軌道(x-axis)
.png")
(二)空白間隔軌道(spacer)
.png")
(三)連續信號軌道(BigWig)
1. 基本信息
1.png")
2. 顏色與透明度
2.png")
3. Y 軸范圍與分辨率
3.png")
4. 缺失值與統計方式
4.png")
5. 信號繪制方式
5.png")
6. 信號運算(實時計算)
6.png")
7. 數據變換
7.png")
8. 文件類型
8.png")
(四)功能區間軌道(BED)
1. 基本信息
1.png")
2. 顏色與邊框
2.png")
3. 顯示方式
3.png")
4. 標簽與字體
4.png")
5. 箭頭與結構參數
5.png")
6. 文件類型
6.png")
(五)基因注釋軌道(GTF)
1. 基本信息
1.png")
2. 基因名稱與轉錄本處理
2.png")
3. 顏色與顯示方式
3.png")
4. 行數、標簽與字體
4.png")
5. 箭頭與結構參數
5.png")
6. 文件類型
6.png")

